

Keeping up with FDA news is not optional if you manufacture, import, label, distribute, or market regulated products in the United States. A single guidance update can change what “complete” looks like for a submission, a new enforcement policy can shift inspection priorities, and a compliance date (like FSMA traceability) can drive major operational work.
This roundup highlights key FDA updates and themes that have mattered recently for regulated companies (with source links), plus practical actions you can take to stay audit-ready and submission-ready.
The FDA updates that tend to matter most (and why)
Most FDA news falls into a few buckets that directly affect regulated businesses:
- New or revised rules (Federal Register) that create legal requirements and hard compliance dates.
- Guidance documents that clarify FDA’s current thinking (not legally binding, but often the standard reviewers and investigators expect).
- Enforcement communications (warning letters, import alerts, recalls) that reveal what FDA is focusing on.
- Process changes (submission formats, portals, required elements) that impact cycle time and acceptance.
A useful habit is to translate every “FDA update” into one of two questions:
- Does this change what we must do, by when?
- Does this change what FDA expects to see, and how they evaluate it?
Cross-industry: organizational and inspection shifts you should track
FDA reorganization and program alignment
FDA has been modernizing its structure and operations, including major changes to the foods program and field/inspection functions. Even when your regulations do not change, organizational changes can affect how quickly issues are triaged, how inspections are prioritized, and where policy decisions sit.
Practical action: If you have open FDA interactions (submissions, compliance follow-ups, import issues), confirm you are using the current contact points and pathways on FDA’s site, and document internal escalation routes for urgent quality or safety events.
Reference: FDA announcements and updates are typically consolidated in FDA Newsroom.
Post-pandemic inspection tools are here to stay
FDA expanded tools such as remote record reviews and alternative assessment approaches during and after the pandemic. Regardless of the exact format, the direction is consistent: your ability to produce clean, consistent, complete records quickly is now a core compliance capability.
Practical action: Run an internal “inspection readiness drill” that tests:
- Batch and lot traceability from raw materials to distribution
- Complaint handling and MDR or adverse event triage (as applicable)
- CAPA effectiveness checks
- Data integrity controls for electronic systems
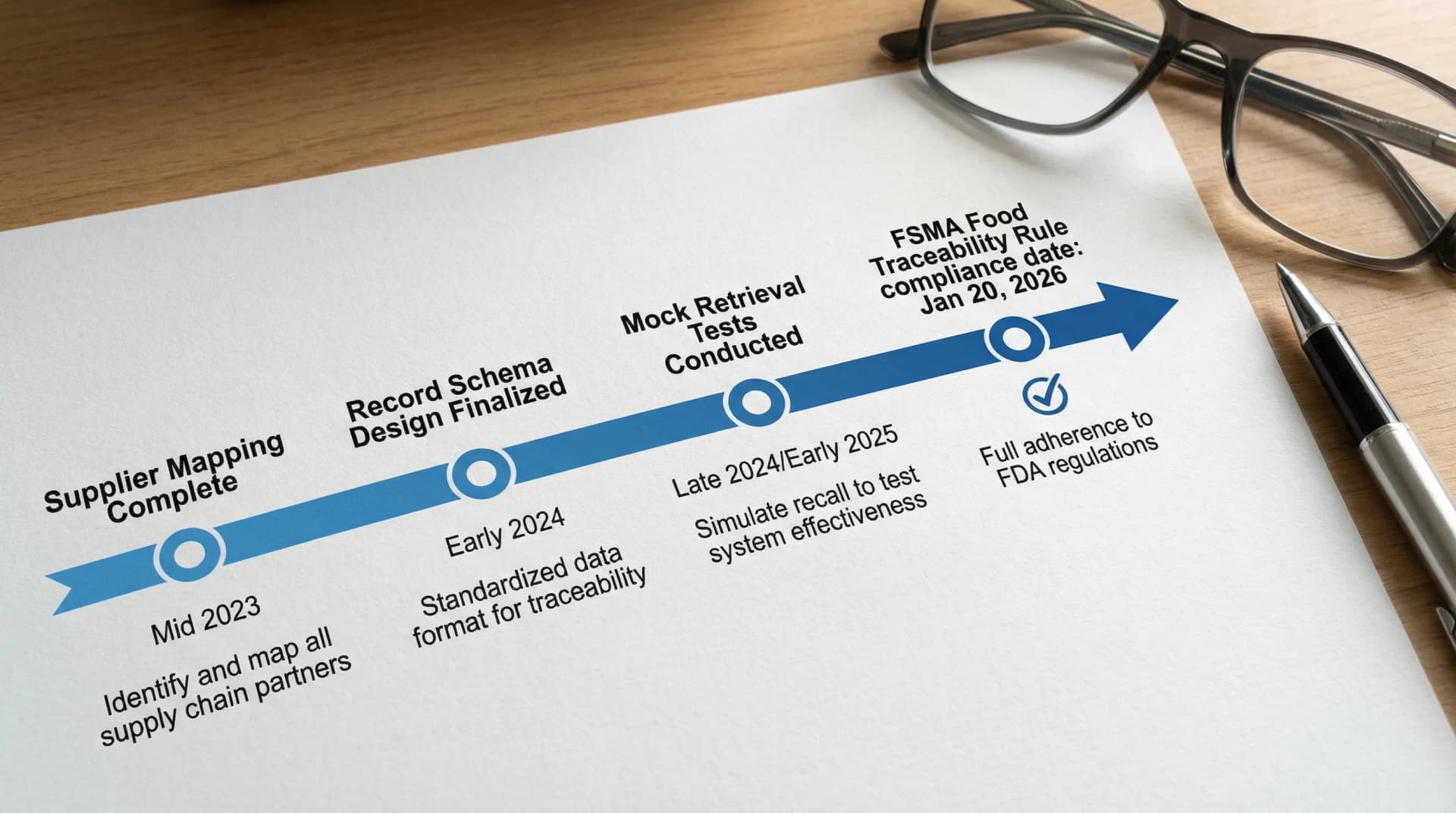
Food: FSMA traceability and “operational readiness” work
Food Traceability Rule compliance date is approaching
One of the most consequential food updates in recent years is the FSMA Section 204(d) Food Traceability Rule, which requires additional traceability records for foods on the Food Traceability List (FTL). FDA’s compliance date is January 20, 2026.
That date matters because traceability is not a “paper policy” project. It typically requires:
- Data standardization across suppliers and internal systems
- Updated receiving, transformation, and shipping records
- Training and clear ownership across operations, quality, and IT
Practical action: If you are in scope, treat 2026 as an operational go-live. Map your current traceability, identify record gaps, and test retrieval times.
Reference: FDA’s overview and resources for the rule are on the Food Traceability Rule page.
Cosmetics: MoCRA implementation continues to reshape compliance
The Modernization of Cosmetics Regulation Act (MoCRA) is the biggest structural change for US cosmetics oversight in decades. Even if your products have been in market for years, MoCRA introduces expectations around registration/listing, adverse event reporting, safety substantiation, and (over time) additional rulemaking that can affect labeling and documentation.
Practical action: Build a MoCRA compliance file that is actually usable during an inspection or inquiry. At minimum, ensure you can produce:
- Facility and product listing status (as applicable)
- Serious adverse event documentation and reporting workflow
- Safety substantiation rationale and supporting materials
- Labeling and claims support files
Reference: FDA’s MoCRA hub is the most reliable starting point for current requirements and updates: MoCRA: Cosmetics.
Medical devices: cybersecurity, eSTAR, and software expectations
Cybersecurity is now a baseline premarket expectation
FDA finalized guidance that outlines cybersecurity expectations across device design controls and premarket submission content. This aligns with broader statutory direction that manufacturers address cybersecurity risk management throughout the product lifecycle.
Practical action: If you have connected devices or any meaningful software component, verify that your quality system and submission templates cover:
- Threat modeling and risk assessment approach
- Secure update mechanisms and vulnerability handling processes
- Documentation reviewers can follow without “tribal knowledge”
Reference: Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions.
eSTAR continues to shape submission mechanics
FDA’s eSTAR is intended to standardize and streamline certain medical device premarket submissions. Even when the underlying scientific expectations are stable, format and completeness requirements can drive refuse-to-accept (RTA) risk and delay timelines.
Practical action: Confirm whether your submission type and center expects or requires eSTAR, and build internal checks so your team does not treat “format” as an afterthought.
Reference: eSTAR (Electronic Submission Template And Resource).
AI/ML-enabled device software: plan for change management
FDA has been developing its approach to AI/ML-enabled device software functions, including documentation expectations for planned updates and change control concepts.
Practical action: If your device includes AI/ML, align engineering, clinical, and regulatory early on what changes are “planned,” how they will be validated, and how they will be communicated to FDA.
Reference: FDA guidance listings for AI/ML and software are best tracked via the Digital Health Center of Excellence.
Drugs and biologics: supply chain integrity and modern trials
DSCSA and serialization readiness remains high impact
The Drug Supply Chain Security Act (DSCSA) continues to drive serialization, verification, and interoperable exchange expectations across the pharmaceutical supply chain. FDA has issued enforcement and “stabilization” communications in recent years to support implementation realities.
Practical action: Treat DSCSA as both a regulatory and partner-management program. Document how you:
- Exchange and reconcile transaction data with trading partners
- Investigate suspect or illegitimate product
- Maintain system validation and data integrity
Reference: FDA’s central resource page is Drug Supply Chain Security Act (DSCSA).
Decentralized clinical trials (DCTs) are getting clearer guardrails
FDA has been publishing guidance and resources on decentralized clinical trial approaches, including the use of local healthcare providers, remote assessments, and digital tools.
Practical action: If you are planning DCT elements, avoid treating “decentralized” as purely operational. Your protocol, monitoring plan, informed consent approach, and data management need to be coherent and inspection-ready.
Reference: FDA guidance documents for clinical investigations can be found through FDA guidance search.
Lab testing and diagnostics: a fast-moving policy area
Clinical lab testing oversight has been an especially dynamic area, including major policy developments affecting lab-developed tests (LDTs). Because this category is sensitive to legal and implementation changes, your team should assume that “what is true today” may shift with litigation, enforcement discretion, and phased timelines.
Practical action: If you develop or rely on LDTs or diagnostic software, establish a monitoring cadence and a decision tree for when you need to update labeling, validation, quality systems, or regulatory pathway assumptions.
Reference: FDA’s medical device policy updates and announcements can be tracked via Medical Devices News and Events.
A practical FDA news monitoring workflow (without drowning in alerts)
Most companies fail at FDA news monitoring in one of two ways: they ignore it until it hurts, or they subscribe to everything and overwhelm the team.
A workable approach is to define “must-track” sources and assign owners.
| Monitoring target | Best for | Where to track | What to do with it |
|---|---|---|---|
| New rules and compliance dates | Legal requirements, deadlines | Federal Register | Update compliance roadmap, budget, and implementation plan |
| New or revised guidance | Reviewer expectations | FDA guidance search | Update submission templates, SOPs, and training |
| Enforcement signals | Inspection and import risk | FDA warning letters | Run gap assessment against cited deficiencies |
| Recalls and safety communications | Post-market safety, vigilance | Recalls, Market Withdrawals & Safety Alerts | Review complaint trending and field action procedures |
To make this operational, set a monthly 30-minute review where regulatory, quality, and operations agree on:
- Which updates create an action item
- Who owns it
- What “done” means (SOP updated, training completed, submission template revised)

When it’s worth bringing in an FDA compliance partner
FDA updates often look simple on the surface (a new guidance, a portal change, a new compliance date) but become complicated when you apply them to your specific product, labeling, quality system, and business model.
Consider expert help when:
- You are entering the US market for the first time and need to avoid preventable submission delays
- Your product category is seeing heightened scrutiny (connected devices, novel claims, complex supply chains)
- You need a US Agent and want alignment between representation, strategy, and submission execution
- You are preparing for an inspection, a major filing, or a due diligence event
CoreQ Advisory supports regulated companies with FDA compliance consulting, US agent services, and end-to-end submission management with fixed-fee transparent pricing. If you want help translating FDA news into concrete regulatory and quality actions, you can learn more at CoreQ Advisory.
CoreQ Advisory is a regulatory intelligence and compliance advisory platform focused on helping manufacturers, importers, and regulated businesses navigate complex U.S. Food and Drug Administration (FDA) requirements.
The CoreQ Advisory editorial team publishes practical guidance covering FDA registrations, Drug Master Files (DMFs), establishment compliance, product identification numbers, and regulatory submission processes. Content is developed through regulatory research, publicly available FDA guidance documents, and industry best practices to provide clear, actionable explanations for companies operating in regulated markets.